La reacción en cadena de la polimerasa (PCR) es una técnica relativamente simple y ampliamente utilizada en el campo de la biología molecular para amplificar y detectar secuencias de DNA y RNA. Comparada con los métodos tradicionales de amplificación y clonación de DNA, los cuales a menudo toman días, la PCR requiere solo unas pocas horas. La PCR es altamente sensible y requiere una plantilla mínima para la detección y amplificación de secuencias específicas. Lo métodos básicos de PCR han tenido grandes avances y hoy en día se puede hacer mucho más que simplemente amplificar fragmentos de DNA o RNA. A continuación se presenta un breve resumen de los principales tipos de PCR y sus diferencias.
PCR
Para la PCR convencional solo se necesita una DNA polimerasa, magnesio, nucleótidos, primers, el DNA molde que se desea amplificar y un termociclador. El mecanismo de la PCR es tan simple como su propósito: 1) El DNA doble cadena (dsDNA) es desnaturalizado (separado en cadenas sencillas) por calor. 2) Los primers se unen a las secuencias compatibles en el DNA cadena sencilla. 3) Los primers son extendidos por la DNA polimerasa (en dirección 5’ – 3’), dando como resultado dos copias de la molécula original de DNA doble cadena. Estas tres etapas comprenden un ciclo de amplificación en la PCR. Cada paso del ciclo debe ser ptimizado por el investigador de acuerdo al DNA molde y el set de primers utilizados. Este ciclo de amplificación se repite usualmente de 20 – 40 veces y el producto amplificado puede ser analizado. La PCR es muy utilizada para amplificar el DNA para su posterior uso en otros experimentos. Esta técnica también tiene aplicaciones en pruebas genéticas o para la detección de DNA patógeno.
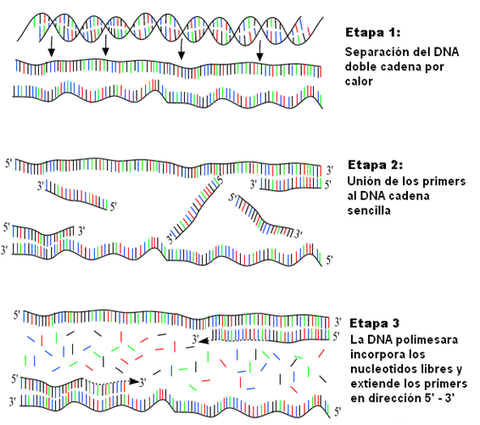
Etapas de un ciclo de amplificación de PCR
Como la PCR es un método altamente sensible y requiere volúmenes de reacción muy pequeños, es recomendable la preparación de una mezcla maestra (master mix) para varias reacciones. La mezcla maestra debe estar bien mezclada y debe dividirse por el número de reacciones, asegurando que cada reacción contenga la misma cantidad de enzima, dNTPs y cebadores (primers). Muchos proveedores ofrecen mezclas de PCR que ya contienen todo, excepto los primers y el DNA molde.
Las regiones con alto contenido de Guanina / citosina (GC) representan un desafío en técnicas de PCR convencional. Las secuencias ricas en GC son más estables que las secuencias con menor contenido de GC (la guanina y la citosina se unen mediante tres puentes de hidrógeno, mientras que la timina y la adenina se unen mediante dos puentes de hidrógeno). Además, las secuencias ricas en GC tienden a formar estructuras secundarias, tales como bucles. Como resultado, las cadenas dobles ricas en GC son difíciles de separar por completo durante la fase de desnaturalización. En consecuencia, la DNA polimerasa no puede sintetizar la nueva hebra sin impedimento alguno. Una temperatura de desnaturalización más alta puede mejorar esto y ajustes hacia una temperatura de unión más alta y un tiempo de unión más corto pueden evitar la unión inespecífica de primers ricos en GC. Ciertos reactivos adicionales pueden mejorar la amplificación de secuencias ricas en GC. El DMSO, el glicerol y la betaína ayudan a interrumpir las estructuras secundarias que son causadas por las interacciones GC y por lo tanto facilitan la separación de las hebras dobles.
HOT START PCR
La amplificación inespecífica es un problema que puede ocurrir durante la PCR. La mayoría de las DNA polimerasas que se utilizan para PCR, funcionan mejor a 68 – 72 ° C. Por lo tanto, la temperatura de extensión elegida debe estar en este rango. Sin embargo, la enzima puede ser también activa en menor grado, a temperaturas más bajas. A temperaturas que están muy por debajo de la temperatura de unión, los cebadores tienden a unirse en forma inespecífica, lo que puede conducir a una amplificación no específica del DNA molde. Esto se puede prevenir usando inhibidores de polimerasa que se disocian de esta sólo cuando se alcanza cierta temperatura. El inhibidor puede ser un anticuerpo que se une a la polimerasa y se desnaturaliza a la temperatura de desnaturalización inicial.
RT-PCR
La PCR de transcripción inversa o RT-PCR, permite el uso de RNA como molde. Un paso adicional permite la detección y amplificación del RNA. El RNA se transcribe de forma inversa en DNA complementario (cDNA) utilizando una transcriptasa inversa. La calidad y pureza del RNA molde es esencial para el éxito de la RT-PCR. El primer paso de la RT-PCR es la síntesis de un híbrido DNA / RNA. La transcriptasa inversa también tiene una función RNasa H, que degrada la porción de RNA del híbrido. La molécula de DNA de cadena sencilla restante sirve entonces como molde para la formación de cDNA, mediante la actividad DNA polimerasa dependiente de DNA, de la transcriptasa inversa. La eficiencia de la reacción de la primera cadena puede afectar al proceso de amplificación. A partir de aquí, se utiliza el procedimiento de PCR convencional para amplificar el cDNA. La posibilidad de revertir el RNA en cDNA por RT‑PCR tiene muchas ventajas. El RNA es monocatenario y muy inestable, lo que dificulta el trabajo con este material. Más comúnmente, sirve como un primer paso en qPCR, que cuantifica la cantidad de RNA que ha sido transcrito en una muestra biológica.


{kind=link}
{kind=link}
{kind=link}
Dejar un comentario